DeNovoMAGIC™
Hybrid


Revolutionizing breeding methods start with sequencing an organism’s DNA to the highest accuracy and unraveling the whole genome assembly. This work fuels informed trait discovery and genomic selection.
NRGene’s DeNovoMAGIC™ is a high-quality de novo assembly solution proven to correctly assemble large, heterozygous and complex genomes correctly. DeNovo assembly means building a reference genome from scratch.
Our technology has revealed the first-ever assemblies of tetraploid heterozygous potato, hexaploid heterozygous sweet potato, octoploid heterozygous strawberry, and the largest crop genome of hexaploid bread wheat.
Based on years of research, development, and optimization by an integrated team of algorithm developers, software engineers, plant geneticists and molecular biologists, DeNovoMAGIC™-Hybrid was developed to be a turn-key solution for churning out new reference genomes in a cost-effective, accurate, and efficient manner.
DeNovoMAGIC™-Hybrid is trusted by dozens of leading scientists from the largest and most prestigious academic institutions and major seed companies world-wide. This technology has made an impact on the genomics world with publications in top-tier academic journals. Our DeNovoMAGIC-Hybrid™ genome assembly software routinely produces new reference genomes for even the most complex, large, heterozygous, and polyploid genomes.
The newest version of DenovoMAGIC™-Hybrid employs a real hybrid assembly to benefit the data quality and unique genome coverage of each: short Illumina reads and long PacBio HiFi reads.
DeNovoMAGIC™- Hybrid is the basis for selecting varieties that perform better than others in yield, disease resistance, climate adaptation, and more.
How it Works
An extensive yet simple tool for analyzing, storing and mining unlimited volumes of DeNovoMAGIC™ delivers the most progressive and novel capability. The only method to truly discover the entire content of the genome!
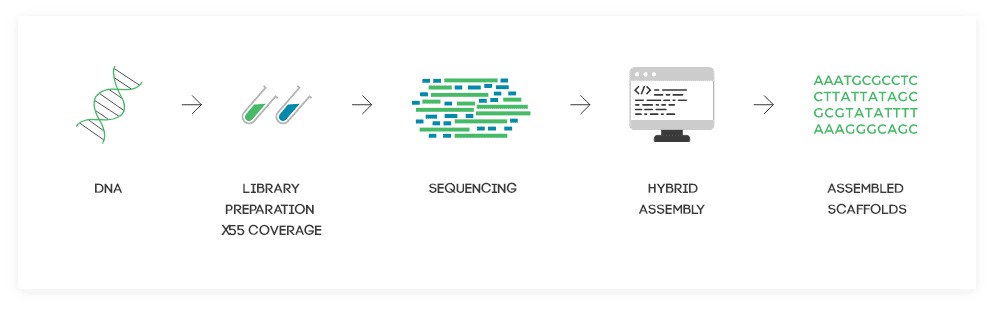
Outstanding Results